Archives
Based on the aforementioned findings
Based on the aforementioned findings, variations of the imidazole heterocycle present in the endogenous ligand histamine (1) led to 2-(thiazol-2-yl)ethanamine (2-TEA, 2) and later to 2-(3-bromophenyl)histamine or 2-(3-(trifluoromethyl)phenyl)histamine (3) (Fig. 3). Interestingly, 2-TEA demonstrated evidently that the tautomeric shift on the nitrogen atoms in imidazole heterocycle is not an important structural element, whereas the finding that 2-TEA displays a moderate efficacy of about 26% of that of histamine with a partial agonist behavior. Noticeably, it has been shown that histamine substitution in the 2-position by a diphenylpropyl moiety could lead to ligands with higher maximal effic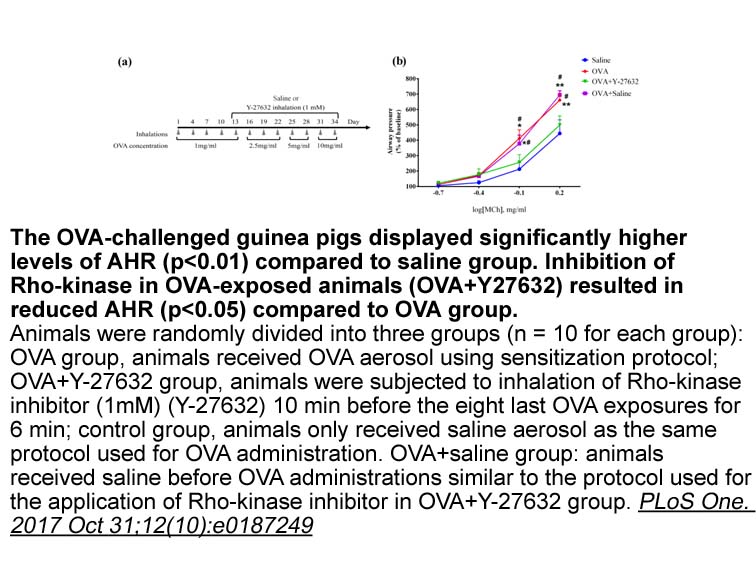 acies than that of histamine (Bruysters et al., 2005, Panula et al., 2015). The latter class, known as histaprodifens, is proposed to simultaneously occupy an agonist as well as an antagonist binding site as demonstrated by site-directed mutagenesis studies and molecular modeling techniques showing slight improvement in efficacy from histaprodifen (4) over to suprahistaprodifen (5) (Bruysters et al., 2005, Jongejan and Leurs, 2005). Noticeably, a distance of three methylene groups between the lipophilic diphenyl structure and the imidazole ring appears to be important, since shorter chains resulted in compounds with partial agonist activity and longer alkyl bridges to antagonists.
acies than that of histamine (Bruysters et al., 2005, Panula et al., 2015). The latter class, known as histaprodifens, is proposed to simultaneously occupy an agonist as well as an antagonist binding site as demonstrated by site-directed mutagenesis studies and molecular modeling techniques showing slight improvement in efficacy from histaprodifen (4) over to suprahistaprodifen (5) (Bruysters et al., 2005, Jongejan and Leurs, 2005). Noticeably, a distance of three methylene groups between the lipophilic diphenyl structure and the imidazole ring appears to be important, since shorter chains resulted in compounds with partial agonist activity and longer alkyl bridges to antagonists.
H1R antagonists
Indeed, it has been the disadvantageous interaction of numerous classical antihistamines with the cardiac human Ether-à-go-go-Related Gene (hERG) potassium alpha-Endorphin that has driven the development of these efficacious drugs. For terfenadine in combination with other CYP3A4 substrates or inhibitors (Bailey et al., 1998) or with liver dysfunction (Kamisako et al., 1995), the plasma levels are highly increased leading to the inhibition of cardiac hERG potassium channels which then may lead to lethal arrhythmias as a result of QT prolongation (Lagrutta et al., 2008, Salata et al., 1995). For instance, terfenadine (14) was eventually withdrawn from the market (Estelle and Simons, 1999) in several, but not all countries, as they were connected with the risk of Torsades de Pointes and sudden death due to binding to hERG of the α-subunit of potassium channels. Also, astemizole was withdrawn in 1999 in U.S and other countries for targeting also hERG channel. Subsequent to the terfenadine removal from the market, it was found that the active zwitterionic metabolite of terfenadine (14), namely fexofenadine (15), although it has a slightly lower affinity for H1R, is devoid of this QT-prolonging side effect and has an enhanced safety profile (Simpson and Jarvis, 2000). Also, it has been revealed that fexofenadine is less sedative, because it displays only a limited brain penetration, possibly due to its affinity for PgP transporters (Molimard et al., 2004, Obradovic et al., 2007). Most of the second generation antihistamines (e.g. cetirizine, 10) demonstrate most of the properties of fexofenadine (15), including the zwitterionic nature. Notably, these ligands show all undoubtedly improved safety profiles and have had coherent therapeutic success.
Histamine H2R
The H2R was recognized in 1972 following the discovery that numerous physiological effects of histamine, including the stimulation of gastric acid secretion, increase of heart rate, and inhibition of rat uterus contraction were not blocked by the H1R antagonist mepyramine (Black et al., 1972). Moreover, cloning of the H2R allowed studies which indicated strong expression in the stomach and brain (Gantz et al., 1991). Clinically, H2R antagonists shaped the treatment regimen of dyspepsia, oesophageal and gastric ulcers, a trend that sustained until these drugs were mostly substituted by proton pump inhibitors (Panula et al., 2015). In addition to the stomach and brain, the H2R is expressed in numerous types of cells, including smooth muscle cells, endothelial and epithelial cells, chondrocytes, neutrophils, eosinophils, monocytes, macrophages, dendritic cells, T and B cells (Jutel et al., 2009, Panula et al., 2015). Interestingly and based on the high resolution crystal structures of bovine rhodopsin, human hH2R was developed by homology modeling (Zhang et al., 2012). The results showed a high quality homology model for hH2R, and three H2R antagonists, namely cimetidine, ranitidine and nizatidine, were applied to the binding site study with this model through molecular docking,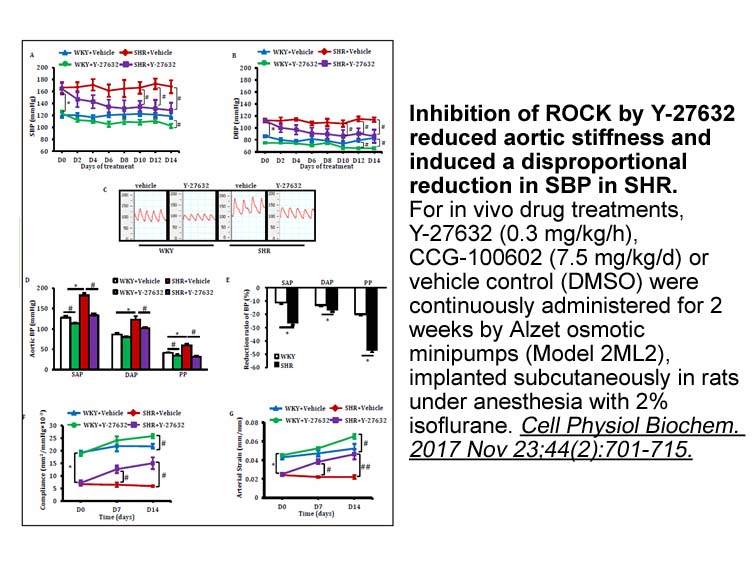 molecular dynamics simulations and molecular mechanics-Poisson-Boltzmann surface area calculations. It has been found that Asp98 in transmembrane domain 7 (TM3) is a major contributor to ligand binding by H-bond interactions. Moreover, Asn159 in TM4 and Asp186 in TM5 were found to be of great importance in stabilizing hH2R-antagonist complexes. Also, two hydrophobic sites especially two residues, namely Val99 in TM3 and Phe254 in TM6, were identified to be essential for their strong hydrophobic interactions with H2R antagonists (Fig. 2B) (Zhang et al., 2012).
molecular dynamics simulations and molecular mechanics-Poisson-Boltzmann surface area calculations. It has been found that Asp98 in transmembrane domain 7 (TM3) is a major contributor to ligand binding by H-bond interactions. Moreover, Asn159 in TM4 and Asp186 in TM5 were found to be of great importance in stabilizing hH2R-antagonist complexes. Also, two hydrophobic sites especially two residues, namely Val99 in TM3 and Phe254 in TM6, were identified to be essential for their strong hydrophobic interactions with H2R antagonists (Fig. 2B) (Zhang et al., 2012).